Diels-alderreactie
De diels-alderreactie is een organische reactie (een cycloadditie), waarbij twee moleculen, een geconjugeerd dieen en een gesubstitueerd alkeen, ook wel het diënofiel genoemd, met elkaar reageren tot een gesubstitueerd cyclohexeen. Deze reactie wordt als een van de krachtigste reacties in de organische synthese gezien, gezien deze op betrekkelijk eenvoudige wijze cyclohexeenringen kan produceren, die van nut zijn in verschillende andere organische synthesen.
De reactie is vernoemd naar de Duitse wetenschappers Otto Diels en Kurt Alder, die in 1950 de Nobelprijs kregen voor de ontdekking van de reactie.
Reactiemechanisme bewerken
De diels-alderreactie is een geconcerteerde reactie; ze vindt plaats in één stap, met slechts één overgangstoestand. Hoge druk of soms verhoogde temperatuur versnellen de reactie. De reactie is een voorbeeld van een pericyclische reactie, meer bepaald een [4+2]-cycloadditie, dit betekent dat er 4 resp. 2 pi-elektronen van de twee reagentia betrokken zijn bij de reactie.
Een schematisch voorbeeld van de diels-alderreactie is de reactie van 1,3-butadieen en etheen. Uit deze reactie ontstaat cyclohexeen.
_bw.svg)
Een orbitalaire verklaring van de reactie wordt weergegeven op onderstaande figuur. In de meeste gevallen is de overlap tussen het HOMO van het dieen en het LUMO van het diënofiel. Hoe kleiner het energieverschil tussen beide orbitalen, des gemakkelijker de reactie verloopt. Om deze reden zijn reacties tussen een dieen gebonden met een elektronenstuwende groep (die de energie van het HOMO verhoogt) en een diënofiel met een elektronenzuigende groep (die de energie van het LUMO verlaagt) doorgaans zeer efficiënt.

Het dieen bewerken
Het dieen gebruikt in de reactie kan verschillende eigenschappen hebben: zowel cyclisch als een open keten en met allerlei substituenten. De enige voorwaarde is dat het dieen in zogenaamde s-cis-conformatie kan aannemen, waarbij de twee dubbele bindingen aan dezelfde kant van de enkele binding staan. 1,3-butadieen kan bijvoorbeeld vrij roteren rond zijn enkele binding, de s-trans-conformatie waarbij de dubbele bindingen om sterische redenen zo ver mogelijk van elkaar af staan is weliswaar stabieler, maar de energiebarrière voor rotatie is klein genoeg om toch de reactieve s-cis-conformatie aan te kunnen nemen. Cyclische diënen die in een vaste s-cis-configuratie staan zijn om die reden uiterst reactief in diels-alderreacties, met cyclopentadieen als typisch voorbeeld. Cyclische diënen in een vaste s-trans-configuratie kunnen dan weer niet reageren in diels-alderreacties.
Het diënofiel bewerken
Het diënofiel beschikt doorgaans over een elektronenzuigende groep (zoals een carbonylgroep) geconjugeerd met het alkeen, of moet met een lewiszuur geactiveerd worden. De eigenschappen van substituenten zullen in hoge mate de regioselectiviteit van de reactie bepalen (zie ook verder).
Hetero-diels-alderreacties bewerken
Zowel het dieen als het diënofiel kunnen heteroatomen bevatten; zij reageren via een hetero-diels-alderreacties. Lawessons reagens kan bijvoorbeeld als diënofiel reageren, waarbij een zesring gevormd wordt. Stoffen met stikstof als heteroatoom zoals imines reageren in een zogenaamde aza-diels-alderreactie en stoffen met zuurstof zoals carbonylgroepen reageren in een oxo-diels-alderreactie.
Selectiviteit bewerken
Hoewel er verschillende isomeren gevormd kunnen worden in een diels-alderreactie, zullen enkele ervan in hogere mate - of zelfs alleen - gevormd worden. Doorgaans beschikt het diënofiel over een elektronenzuigende groep, en het dieen over een elektronenstuwende groep, beiden in conjugatie. Aan de hand van resonantie kunnen partiële ladingen toegewezen worden in de respectievelijke reagentia, waardoor men de relatieve oriëntatie van het dieen en diënofiel tijdens de reactie kan voorspellen, en zo ook het welke structuurisomeer gevormd wordt.
Op volgend figuur wordt aangetoond waarom van de gegeven dieen (links) en diënofiel (rechts) slechts één structuurisomeer gevormd wordt (de ortho-variant) en niet de andere mogelijkheid (de meta-variant).

Het cis-principe bewerken
Volgens het cis-principe of de alder-steinregels blijft de relatieve stereochemie van de substituenten in de reagentia behouden. Dit betekent dat als een diënofiel twee substituenten heeft die cis staan tegenover elkaar, ze in het reactieproduct nog steeds cis zullen zijn. Hetzelfde geldt voor trans-isomeren van het diënofiel, en ook voor de relatieve configuratie van substituenten van het dieen. Onderstaande reactie toont als voorbeeld dit behoud van configuraties:

De endo-regel bewerken
Het cis-principe verklaart hoe de relatieve configuratie van substituenten op het dieen of diënofiel behouden zal blijven, maar niet hoe die van het dieen en het diënofiel relatief ten opzichte van elkaar zullen terechtkomen. Echter blijkt dat als het diënofiel beschikt over substituenten met pi-elektronen (zoals bijvoorbeeld een carbonylgroep) een bepaald product meer wordt gevormd, namelijk dat waarbij substituenten die van het diënofiel komen in de richting van het de nieuwe dubbele binding wijzen.
Het product waarbij substituenten van het diënofiel in de richting van de dubbele binding wijzen noemt men het endo-product, het andere is het exo-product. Het endo-product wordt in bepaalde gevallen dus meer gevormd: dit is de endo-regel. De mogelijke benaderingen van een diënofiel in termen van endo- en exo-isomeren worden hieronder aangehaald:

De reden hiervoor ligt in de overgangstoestand in de reactie die leidt tot het endo-product. Als het diënofiel een oriëntatie ten opzichte van het dieen aanneemt die leidt tot het endo-product, kan er extra overlap optreden tussen de pi-orbitalen van de substituenten van het diënofiel, en de andere pi-orbitalen van het dieen. Deze extra overlap gedurende de overgangstoestand verlaagt de energie ervan, en dus ook de activeringsenergie van de reactie. Om die reden zal het endo-product sneller gevormd worden dan het exo-product. Op onderstaande figuur wordt dit toegelicht met een klassiek voorbeeld: de reactie tussen cyclopentadieen en maleïnezuuranhydride. Links is de endo-oriëntatie te zien, die extra stabiliserende overlap toelaat, terwijl bij de exo-oriëntatie rechts alleen overlap tussen de orbitalen die leiden tot de nieuwe bindingen mogelijk is.

Vaak is echter om sterische redenen het exo-product stabieler is dan het endo-product, en kan men met thermodynamische reactiecontrole (omstandigheden waarbij het meest stabiele product met meest gevormd wordt, en niet het product dat het snelst kan gevormd worden) meer van het exo-product verkrijgen. De omgekeerde reactie, waarbij uit een cyclohexeen een dieen en een alkeen ontstaan, wordt een retro-diels-alderreactie genoemd. Onderstaande afbeelding toont van een zeker product zowel het endo-product (links) en het exo-product (rechts).

Toepassingen bewerken
Diels-Alder-reacties worden gebruikt in de synthese van verschillende chemische producten. Een interessant voorbeeld zijn de gechloreerde insecticiden aldrin en dieldrin, genoemd naar de ontdekkers van de reactie. Aldrin wordt gevormd door hexachloorcyclopentadieen met norbornadieen in een diels-alderreactie met elkaar te laten reageren. Een epoxidatie van de norborneenring vormt hieruit dan dieldrin.
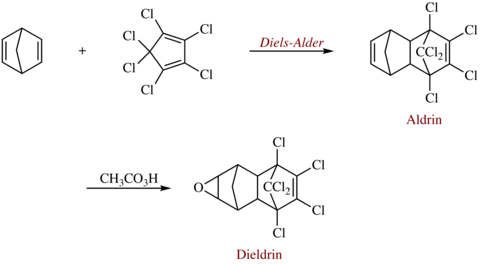
Synthese van aldrin en dieldrin via een diels-alderreactie.

Ook het insecticide en acaricide endosulfan, synthetische geurstoffen als Phenoxanol® (3-methyl-5-fenylpentanol) en de chemische grondstof antrachinon worden onder andere met een diels-alderreactie gesynthetiseerd.